{kind=link}
{kind=link}
A robust experimental and computational analysis framework at multiple resolutions, modalities and coverages
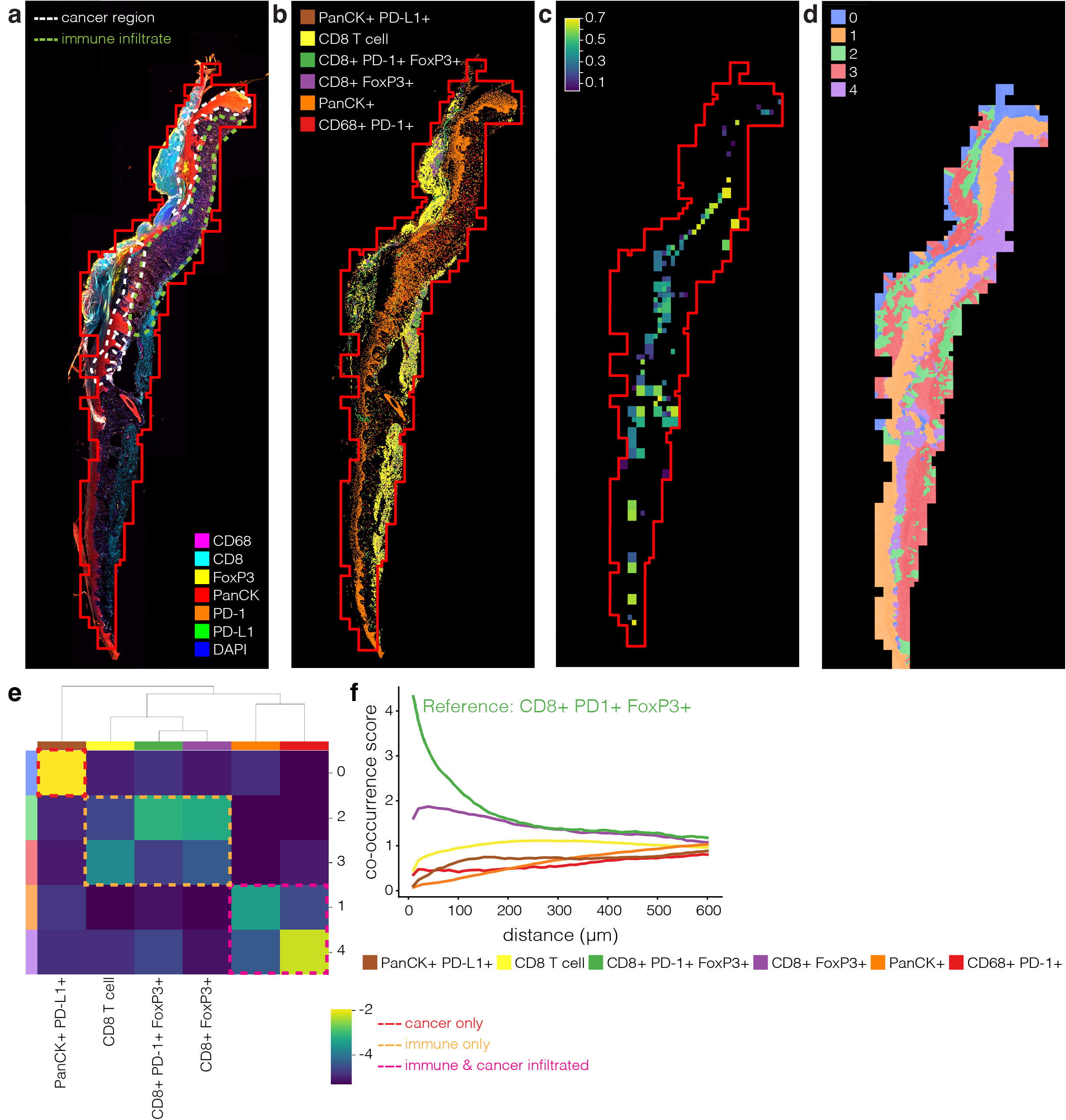
This paper is about framework called STRISH to study cell-cell interaction through ligand-receptor

| PhD Candidate | Oct 2019 - Present |
| 1. Conduct research and analysis for cellular communication within spatial context in cancer using imaging and sequencing data | |
| 2. Propose a novel computation package called STRISH to identify cells colocalisation with ligand-receptor throughout the tissue using spatially resolved multi-omics data (spatial transcriptomic and proteomic data) | |
| 3. Adapt the spatial analyses (i.e. Delaunay triangulation, K nearest neighbors, Ripley’s K function) and heterogeneity scoring measurements (i.e. Shanon entropy, Rao’s quadratic entropy, graph modularity) to identify the spatial differences across cancer subtype | |
| 4. Collaborate with other research groups to develop and conduct the analysis for scRNA-seq, ATAC-seq and spatial -omic data | |
| 5. Tutor course introduction to Bioinformatic BINF6000 | |
| Bioinformatic Scientist | Mar. 2019 - Oct. 2019 |
| 1. Design the workflows to analyse workflow to analyse DNA sequencing data to build the largest population-scale biomedical database for Vietnamese genome | |
| 2. Develop API to automate data QC process and upload meta data to the genomic variant database for Vietnamese population | |
| 3. Benchmark for accuracy and speed of the performance of variant calling platforms including Dragen (Illumina), DeepVariant (Google), and GATK | |
| AI Engineer | Oct. 2018 - Mar. 2019 |
| 1. Develop backbone of document analysis solution using machine learning (ML) and deep learning (DL) to extract information from contracts | |
| 2. Leverage state-of-the-art NLP transformers BERT as the alternative solution for information extraction models to deliver higher accuracy 90%+ overall and F1 score at 0.86 with limited training data (approx. 100 samples) | |
| Visiting Scholar | Mar. 2018 - Sep. 2018 |
| 1. Chromosome conformation capture techniques (Hi-C) provide a wealth of data on the three-dimensional architecture of genomes | |
| 2. Introduce dimensional reduction including Autoencoder, PCA and SVG and robust differential analysis for noisy and high dimensional Chromosome 3D maps | |
| 3. Autoencoder achieved higher true positive value compared to PCA by 15%-30% proportion to the size of consideration block |
Programming Languages: Python, R, Bash Shell, JavaScript, Java, C/C++, SQL, LATEX
Developer Tools: Git, Docker, Anaconda, Slurm, PBS
Framework and Tools: sklearn, pandas, skimage, scipy, SimpleITK, tensorflow, Keras, QuPath, CellProfiler, napari, Seurat, scanpy
This paper is about framework called STRISH to study cell-cell interaction through ligand-receptor
Here, we benchmark data from Beijing Genomics Institutes (BGI) DNBSEQ-G400 low-cost sequencer against data from a standard Illumina instrument (HiSeqX10). For comparisons, the same bulk ATAC-seq libraries generated from pluripotent stem cells (PSCs) and fibroblasts were sequenced on both platforms.
This paper combines pairwise constraint to unsupervised clustering algorithm to produce a semi-supervised algorithm called CNN-PC
Here, we conducted a microarray analysis of 13 women affected by MRKH syndrome, resulting in the identification of chromosomal changes, including the deletion at 17q12, which contains both HNF1B and LHX1. We ablated Hnf1b specifically in the epithelium of the Müllerian ducts in mice and found that this caused hypoplastic development of the uterus, as well as kidney anomalies, closely mirroring the MRKH type II phenotype. Our results support the investigation of HNF1B in clinical genetic settings of MRKH syndrome and shed new light on the molecular mechanisms underlying this poorly understood condition in women’s reproductive health.
We introduce a convolutional autoencoder an unsupervised machine learning technique to produce a similarity function to compare areas between pairs of Hi-C matrices
Using Spatial Transcriptomics technology, we develop a user-friendly deep learning software, SpaCell, to integrate millions of pixel intensity values with thousands of gene expression measurements from spatially barcoded spots in a tissue.
Using Hyperion Imaging Mass Cytometry (IMC), we simultaneously profiled 16 protein markers for each tissue section, capturing molecular signatures of tissue architecture, cancer cells, and immune cells. In this project we aim to capture tissue morphology, cancer cell types, multi-parameter protein contents of single cells in within morphologically intact tissue sections of colorectal tumours from 52 patients.